
- Père 175 cm, mère 168 cm.
Pas d’ antécédents familiaux.
Né à 40 semaines à 3300 g et 50 cm. - Taille 50 cm, poids 3 300 g.
Bourgeon génital de 2 cm avec hypospade périnéal.
2 testicules (15×10 mm) dans le scrotum.
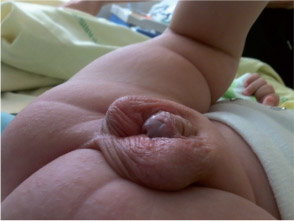
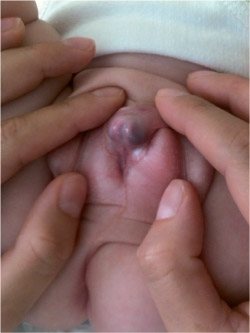
Quelles sont vos hypothèses ?
Cliquez sur les flèches blanches pour accéder à la suite du cas clinique.
Ce n’est pas une forme « classique » de déficit en 21-hydroxylase présentent à la naissance une virilisation variable de leurs OGE: hypertrophie du clitoris, fusion des grandes lèvres, absence de l’orifice vaginal au périnée, urètre pénien, classés en cinq stades de Prader.
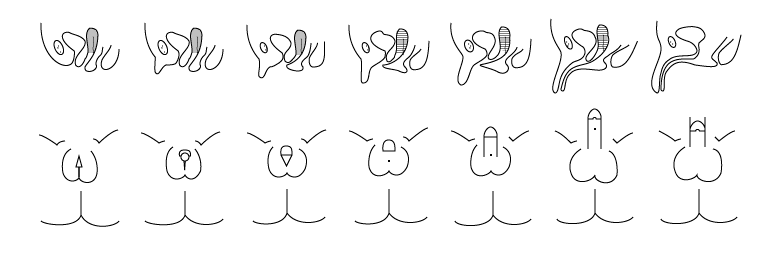
S’il était une fille masculinisée, Claude aurait été classé Prader 3 ou 4. Mais des testicules étant palpés dans le scrotum, le caryotype comporte un chromosome Y.
Le cas de Claude peut donc être classé, très probablement, comme un DSD 46,XY
Les DSD 46,XY sont caractérisés par des OGE anormaux, en raison d’une virilisation incomplète intra-utérinepar insuffisance de production de la testostérone ou résistance aux androgènes. Les causes de DSD 46,XY entrent dans 4 catégories :
Selon l’étiologie, des structures müllériennes sont ou non présentes.
Quel est le seul diagnostic possible pour Claude ?
Voici une liste d’anomalies de la différenciation sexuelle :
Les troubles de la détermination et du développement fœtal des testicules
Les DSD 46,XY associés à des manifestations syndromiques qui ne sont pas à discuter chez Claude.
Les DSD 46,XY dus à des mutations perte de fonction du gène LH/CGR et à l’altération de la différenciation des cellules de Leydig (forme partielle)
Les mutations homozygotes ou hétérozygotes composites perte de fonction sont associées à des OGE féminins. Les patients avec des formes plus modérées ont des OGE masculins avec micropénis et/ou hypospade ou une infertilité sans OGE anormaux. La LH est souvent élevée. Les cellules de Leydig sont absentes ou rares (hypoplasie des cellules de Leydig).
Les DSD 46,XY par défauts de production de la testostérone
Les défauts de la stéroïdogenèse testiculaire
Les DSD 46,XY par déficit en 5α-réductase 2 (5RD2)
C’est un diagnostic possible pour Claude. Le gène SRD5A2 pour la 5α-réductase de type 2. Les patients 46,XY atteints ont en général des OGE presque féminins, une verge très courte pouvant être prise pour un clitoris, et une régression Müllérienne. Les testicules sont généralement inguinaux. A la naissance, les taux de dihydrotestostérone (DHT) sont bas. A la puberté, lorsque l’activité de l’isoenzyme de type 1 augmente, une virilisation se produit. Le diagnostic de déficit en 5α-réductase est évoqué lorsque le rapport T/DHT est > 10 mais un rapport T/ DHT au cours de la période néonatale > 8,5 peut être en faveur du diagnostic. 33 mutations différentes (dont 20 dans des familles consanguines), ont été retrouvées chez 55 patients.
Les défauts de l’action des androgènes sont la cause la plus fréquente de DSD 46,XY
Ce diagnostic n’est pas plausible chez Claude, car il entraîne généralement une petite verge, un scrotum fusionné, et une cryptorchidie. L’insensibilité aux androgènes est complète quand il y a une absence complète d’action ou partielle (PAI) quand il existe des degrés variables de défauts d’action. Le phénotype est très variable et la confusion avec d’autres causes de DSD 46,XY est fréquente.
Un bref commentaire concernant le choix de sexe des enfants DSD 46,XY
Un nouveau-né qui présente des organes génitaux anormaux doit être considéré comme une urgence médicale en raison des problèmes vitaux présents dans certains cas. Une analyse doit être lancée immédiatement pour tenter d’obtenir un diagnostic précis lorsque cela est possible. Un choix de sexe masculin ou féminin peut être discuté. Le sexe masculin est recommandé pour les patients atteints de déficit en 5α-réductase ou en 17β-hydroxystéroïde déshydrogénase, en raison du potentiel de fertilité masculine, malgré un phénotype néonatal parfois très féminin.
Après le diagnostic, les parents doivent être informés sur le DSD, y compris sur les conséquences à long terme. Une fois que les parents ont reçu cette information, l’équipe médicale doit les soutenir activement dans le choix de sexe qui sera fait avec eux.
Premiers résultats biologiques de Claude

Quel est votre diagnostic ?
Seul le déficit en 3β-hydroxystéroïde déshydrogénase (HSD3B2) peut expliquer l’association de l’hypospade et de la valeur de 17OHP chez un nouveau-né. C’est une forme très rare d’hyperplasie congénitale des surrénales.
Claude a été traité par hydrocortisone et fludrocortisone pour éviter la perte de sel.
L’enzyme convertit les stéroïdes Δ4 en Δ5: la prégnénolone en progestérone, la 17OHprégnénolone en 17OH-progestérone, et la DHEA en androsténedione.
Elle assure aussi une voie de synthèse alternative de la testostérone à partir de l’androstenediol dans les testicules.
Le déficit en 3ß-HSD altère la stéroïdogenèse dans les gonades et les surrénales et la production des stéroïdes sexuels, en plus du cortisol et de l’aldostérone.
Les cellules de Leydig expriment la 3ß-HSD dés 18 semaines vie intra-utérine.
Les formes d’HCS avec perte de sel nécessitent une reconnaissance et un traitement rapides.
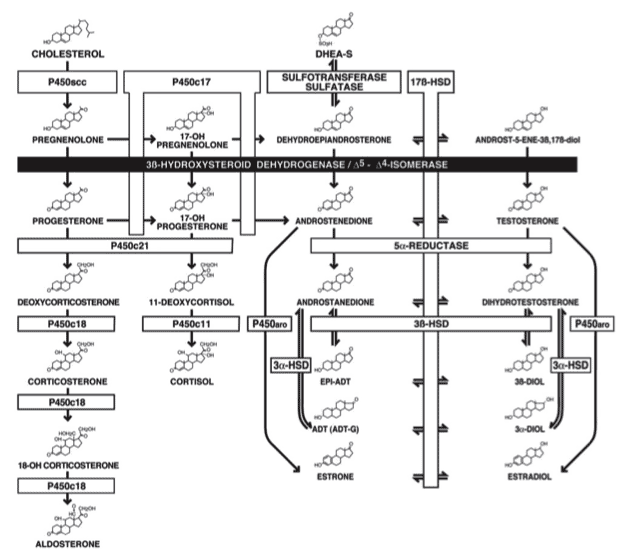
Phénotypes de déficit en 3ß-HSD dans la littérature
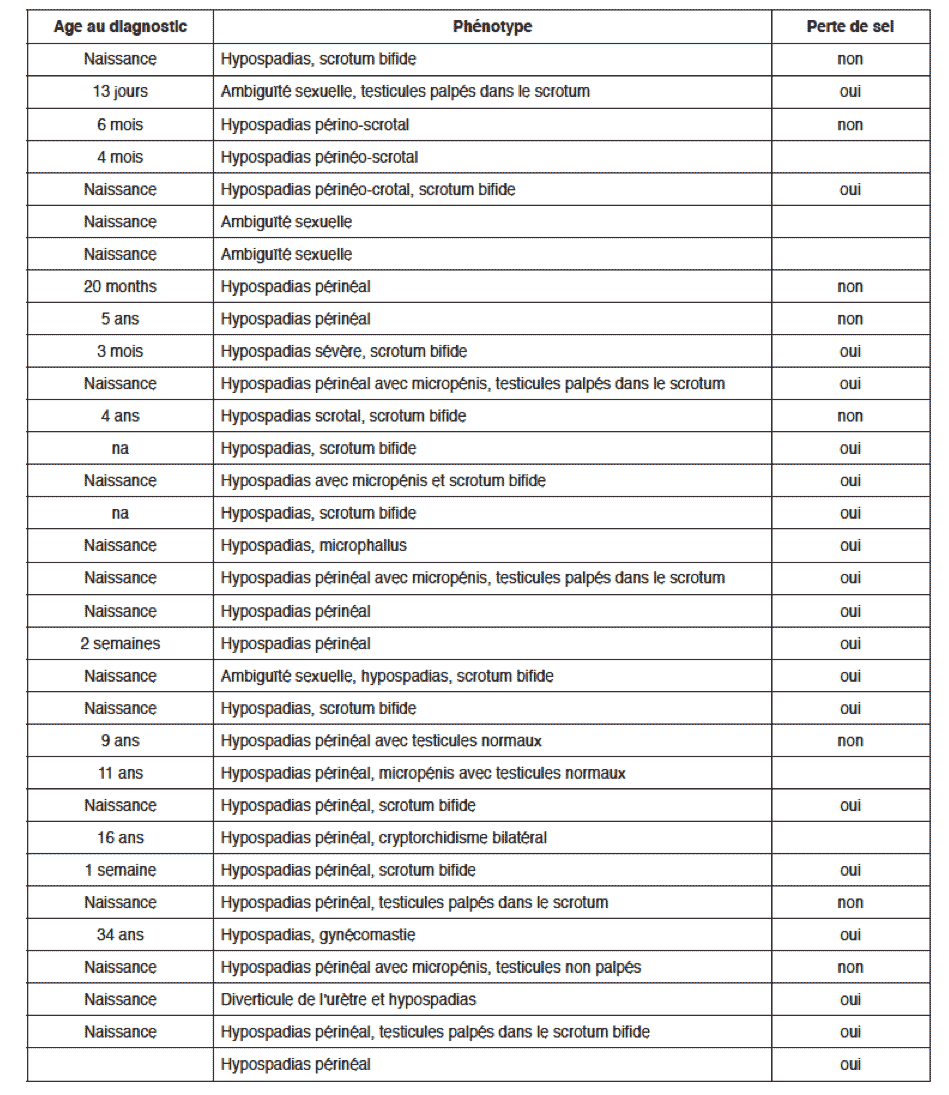
Les garçons porteurs de déficit en 3ß-HSD présentent un hypospadias périnéal ou périnéoscrotal comme indiqué dans le tableau précédent.
Chez les filles, l’inhibition complète ou partielle de l’activité 3ß-HSD dans les surrénales et les ovaires n’est pas accompagnée par un changement notable de la différenciation des OGE (il existe parfois une petite hypertrophie clitoridienne).
La raison de cette différence frappante entre le phénotype des individus masculins et féminins est que la carence en 3ß-HSD abaisse les niveaux de testostérone au-dessous des niveaux requis pour le développement normal des OGE.
La forme classique avec perte de sel est diagnostiquée au cours des premiers mois de vie en raison de la perte de sel, Elle peut être mortelle si elle n’est pas diagnostiquée et traitée rapidement.
Parce que la différenciation sexuelle est normale chez les nouveau-nés de sexe féminin affectés par un déficit en 3ß-HSD sans perte de sel, le diagnostic n’est porté que dans les pays qui pratiquent le dépistage néonatal du 21OHD.
Le diagnostic hormonal
Prégnénolone, 17OHprégnénolone, et DHEA sont élevés.
Le meilleur critère diagnostique est un 17OH-prégnénolone > 100 nmol/l après stimulation par l’ACTH. Lamesure de la 17OH-prégnénolone doit être effectuée, quand un niveau élevé de 17OHP est observé chez un un clitoris un peu trop gros.
Le diagnostic moléculaire
A ce jour, 37 mutations (dont cinq du cadre de lecture, quatre non-sens, une délétion, une mutation d’épissage, et 26 mutations faux-sens) ont été identifiées dans le gène HSD3B2 chez 60 personnes, de 47 familles, porteuses de déficit en 3ß-HSD de type II.
A la génitographie, Claude avait une grande cavité postérieure et un hypospade périnéal.
La taille du pénis a été normalisée avec des injections de testostérone jusqu’à une taille de 35 mm.
Une génitoplastie a été effectuée à l’âge de 7 mois.
Message résumé
Même lorsque les testicules sont palpés dans le scrotum d’un bébé ayant un hypospade, la 17-OH-progestérone doit être mesurée. Ceci est vrai lorsqu’un dépistage néonatal n’a pas été réalisé.
Le défaut de virilisation des garçons est la conséquence d’une synthèse de testostérone diminuée dans les glandes surrénales et les testicules. Bien que la DHEA soit élevée, c’est un androgène faible, et trop peu de testostérone est produite dans le foie pour compenser l’insuffisance de testostérone testiculaire. Le degré de virilisation est variable, de modéré à sévère.
L’hypospade est réparé chirurgicalement, les testicules abaissés dans le scrotum s’ils n’y sont pas, et la testostérone prescrite, si nécessaire, à la puberté.




